SMR简介以及代码实操
一、简介
SMR是由西湖大学杨健教授团队开发并支持的一个进行SMR以及HEIDI检验的软件工具。其官方网址是SMR,其中包含了对SMR的详细介绍以及操作教程。
SMR(Summary data-based Mendelian Randomization analysis),其核心思想就是利用eQTL数据对某些phenotype进行MR分析,得到阳性结果后,因为eQTL本身对基因表达的影响,得到结果:基因表达影响phenotype。在官网里面也放出了相关的文献,文章地址,建议一读,能增加对SMR本身的理解。
下面是SMR的操作步骤:
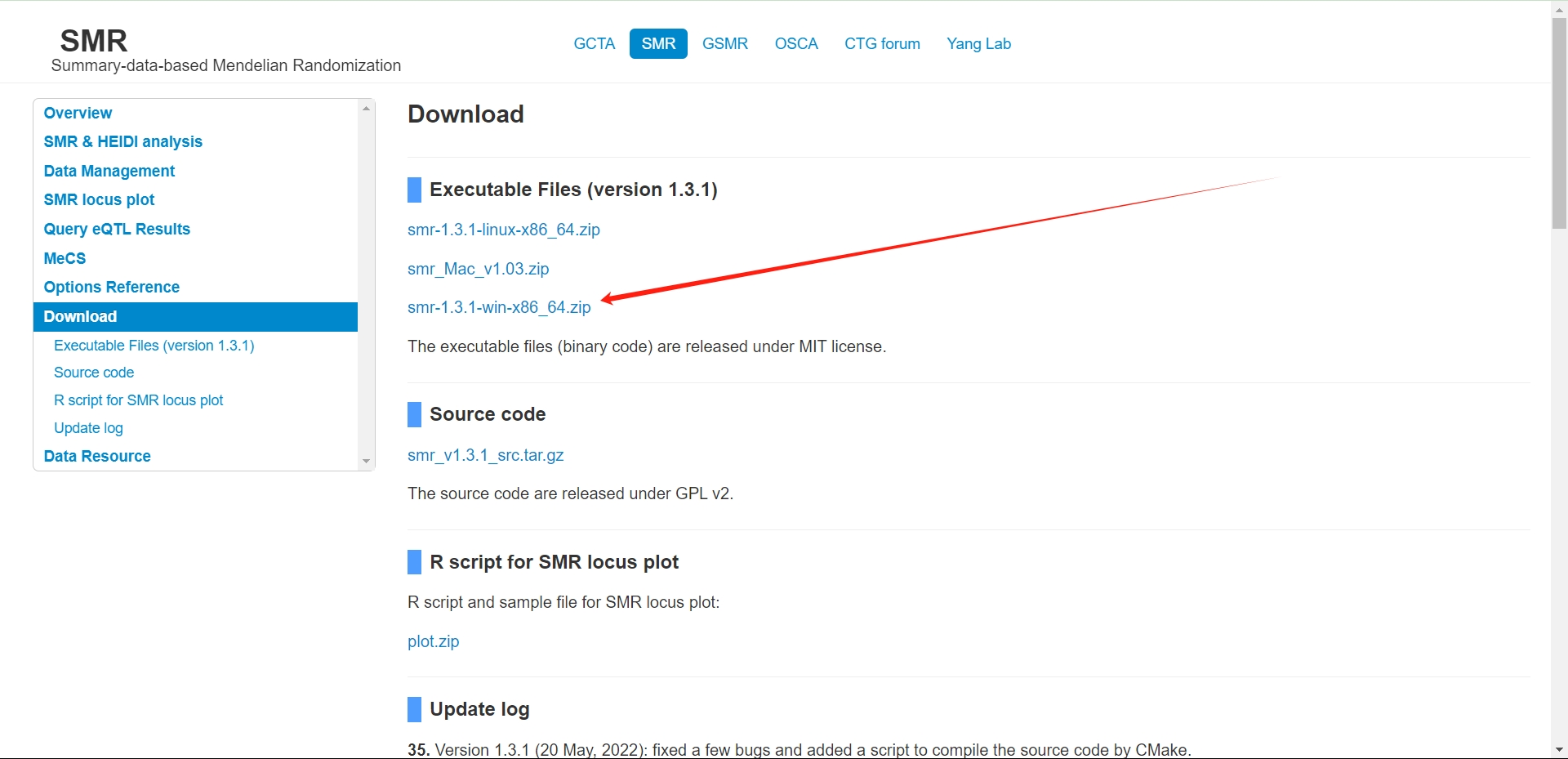
预备步骤1:软件下载
- 进入SMR网站后,在Download里面选择下载。这里我使用Windows版本进行演示,win版本操作简单,对于不会Linux的人来说非常友好,所以下载win版本

- 下载好之后选择一个合适的位置解压即可,解压后应该能看到下面这三个文件。其中.exe后缀的就是我需要使用的SMR软件,但是这是一个命令行工具是不能直接打开的。
预备步骤2:数据准备
- 在完成SMR软件下载之后,我们需要准备我们分析所需要的数据。其中包括:GWAS数据以及eQTL数据。
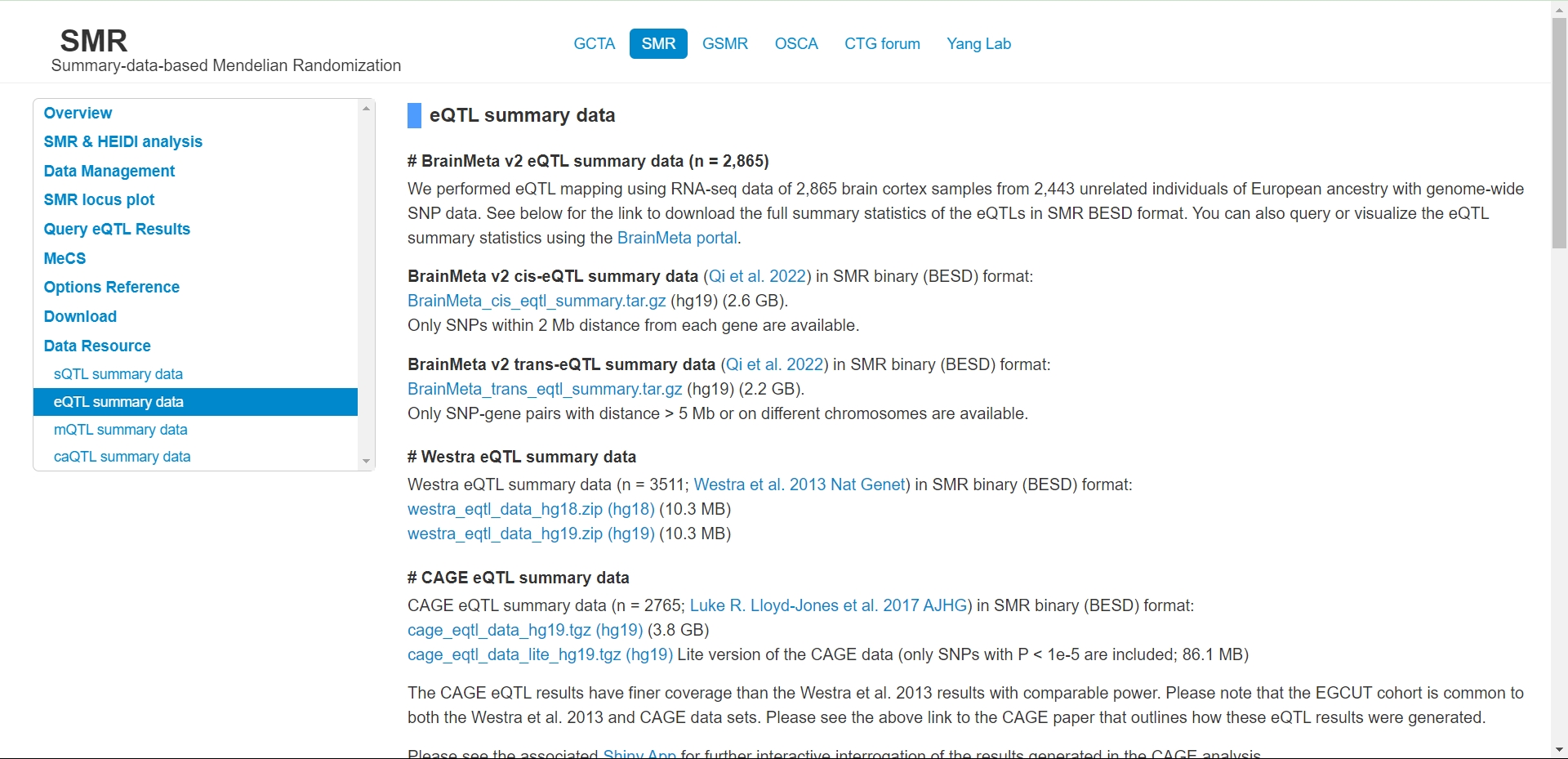
eQTL数据准备 - eQTL数据在SMR官方网站提供了很多数据量很大的数据。下载速度很快,分析者可以按照自己的需求进行选择。eQTLGen也提供了SMR格式的血液样本的eQTL数据。
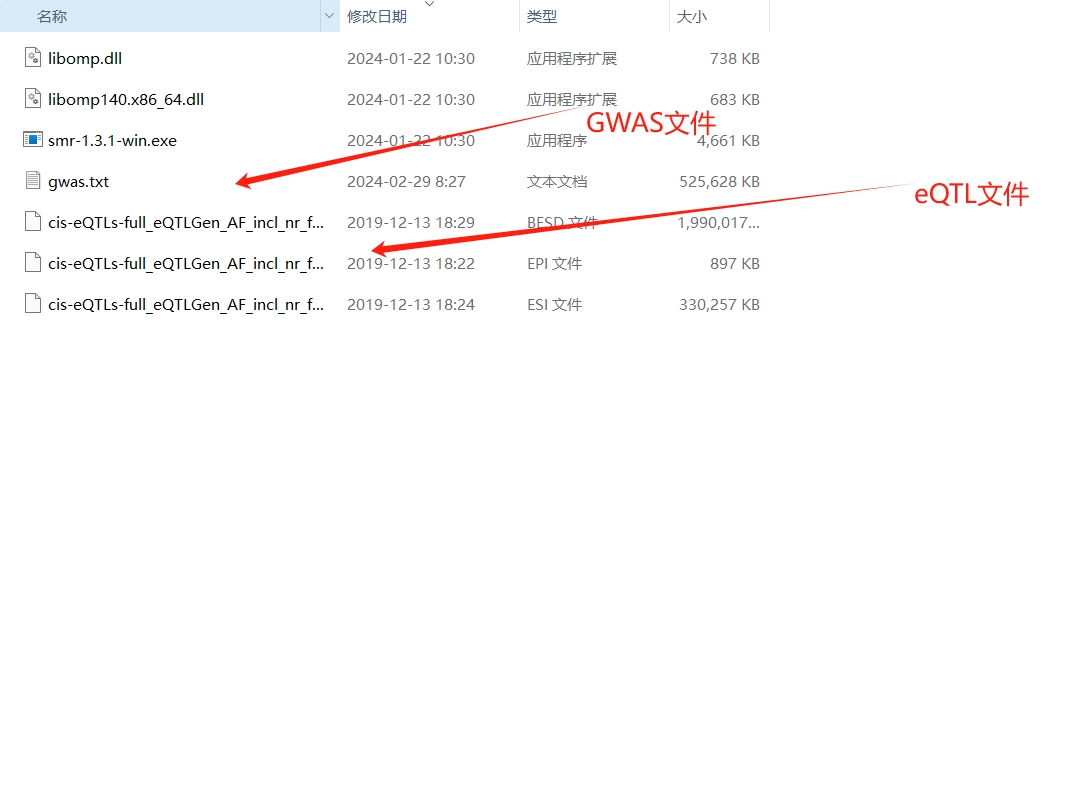
- 我们下载好之后会发现,eQTL数据分别是由三个文件组成,包括.BESD文件 .EPI文件 .ESI文件。将其与SMR程序放到同一个文件夹中,如下图所示(以外周血eQTLGen为例)。这个就是SMR格式的eQTL文件,后面直接分析就可以了。
GWAS数据准备
-eQTL数据准备好之后,我们继续准备GWAS数据。GWAS数据在做孟德尔随机化分析的时候,大家往往直接在TwoSampleMR包通过IEU的API在R中直接读取,这里因为是本地分析,所以需要下载GWAS数据并转换为TXT文件进行分析。 - 我们下载IEU opengwas的数据举例,IEU opengwas的数据格式是vcf格式,也是大多数GWAS数据的常用格式。
- 我选择了eczemas,也就是湿疹的GWAS数据进行处理,处理的过程也是非常简单。几行代码就可以搞定。代码附上。
setwd("D:\\MR\\SMR\\Blog") library(VariantAnnotation) library(gwasglue) gwasdata<-readVcf("ieu-a-996.vcf.gz") gwasdata<-gwasvcf_to_TwoSampleMR(gwasdata,"outcome") gwasdata<-data.frame(gwasdata$SNP, gwasdata$effect_allele.outcome, gwasdata$other_allele.outcome, gwasdata$eaf.outcome, gwasdata$beta.outcome, gwasdata$se.outcome, gwasdata$pval.outcome, gwasdata$samplesize.outcome) colnames(gwasdata)<-c("snp","A1","A2","freq","b","se","p","n") write.table(gwasdata,"gwas.txt",quote = FALSE,row.names = FALSE)完成上述步骤之后,我们就得到自己的GWAS文件了,同样的也是和SMR程序放到同一个目录下。
LD文件准备
- 除GWAS数据以及eQTL数据以外,还需要参考基因组文件,这里我是使用的网上的资源的,应该是一个IEU的服务器提供的,这里附上下载链接,LD文件下载大家也可以直接去1000 Genome Projects的官方网站下载,目前笔者还没搞清楚怎么使用,后期弄明白了也许会出一个教程。毕竟这个项目还是很重要的。
- 下载好之后,里面应该是包含多个人种的数据,一般使用欧洲人种的数据,需要将解压的数据,放到一个特定的路径下(不需要放到SMR程序路径下,避免影响其他分析使用LD文件)
我的LD文件路径是D:\EUR_ref\1kg.v3\EUR需要使用EUR时,就在路径的最后加上指定的人种,如果使用亚洲人群的话,那么路径就是D:\EUR_ref\1kg.v3\EAS。
数据准备完成!!现在SMR程序文件下应该包含如下文件,请自查
SMR分析
1. win+R输入cmd,打开终端命令行
因为SMR是一个没有图形化界面的软件,所以需要直接使用命令行窗口进行使用。win键+R输入cmd,可以快速打开命令行窗口
2. 切换到SMR程序目录
D:由于不能跨盘符切换目录,首先切换到D盘,cd D:\MR\SMR\smr-1.3.1-win-x86_64\smr-1.3.1-win-x86_64切换到指定目录,输入smr-1.3.1-win.exe回车之后就进入SMR软件了。
3. SMR分析
按照官网给的教程,改成我们自己的参数和数据,直接进行分析。
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --out mysmr --thread-num 10 - smr 这里官网上写的smr,个人实测应该得用程序名,我的压缩包里面的程序名是
smr-1.3.1-win.exe - bfile 个人的LD文件路径,我的是,D:\EUR_ref\1kg.v3\EUR
- gwas-summary mygwas.ma ---gwas文件名
- beqtl-summary myeqtl --eqtl文件名(无后缀,三个文件)
- thread-num 指定 OpenMP 线程数 并行计算。默认值为 1。个人理解可能是和计算机计算相关的参数。这里按照网上资料,使用10
- out mysmr --SMR结果文件
简单的修改之后就得到了自己使用的命令了
smr-1.3.1-win.exe --bfile D:\EUR_ref\1kg.v3\EUR --gwas-summary gwas.txt --beqtl-summary cis-eQTLs-full_eQTLGen_AF_incl_nr_formatted_20191212.new.txt_besd-dense --out SMR_outcome --thread-num 10 --diff-freq-prop 0.92
diff-freq-prop 参考知乎上一位博主的教程,知乎链接,他设置了diff-freq-prop就是基因频率的阈值,这一步大家可以按照自己的需求去做,或者多换几种不同的阈值进行尝试。之后关于这个参数我有新的发现会继续更新。




上述的准备工作做好之后,我们可以直接回车进行分析啦!整个过程大概8-9分钟。
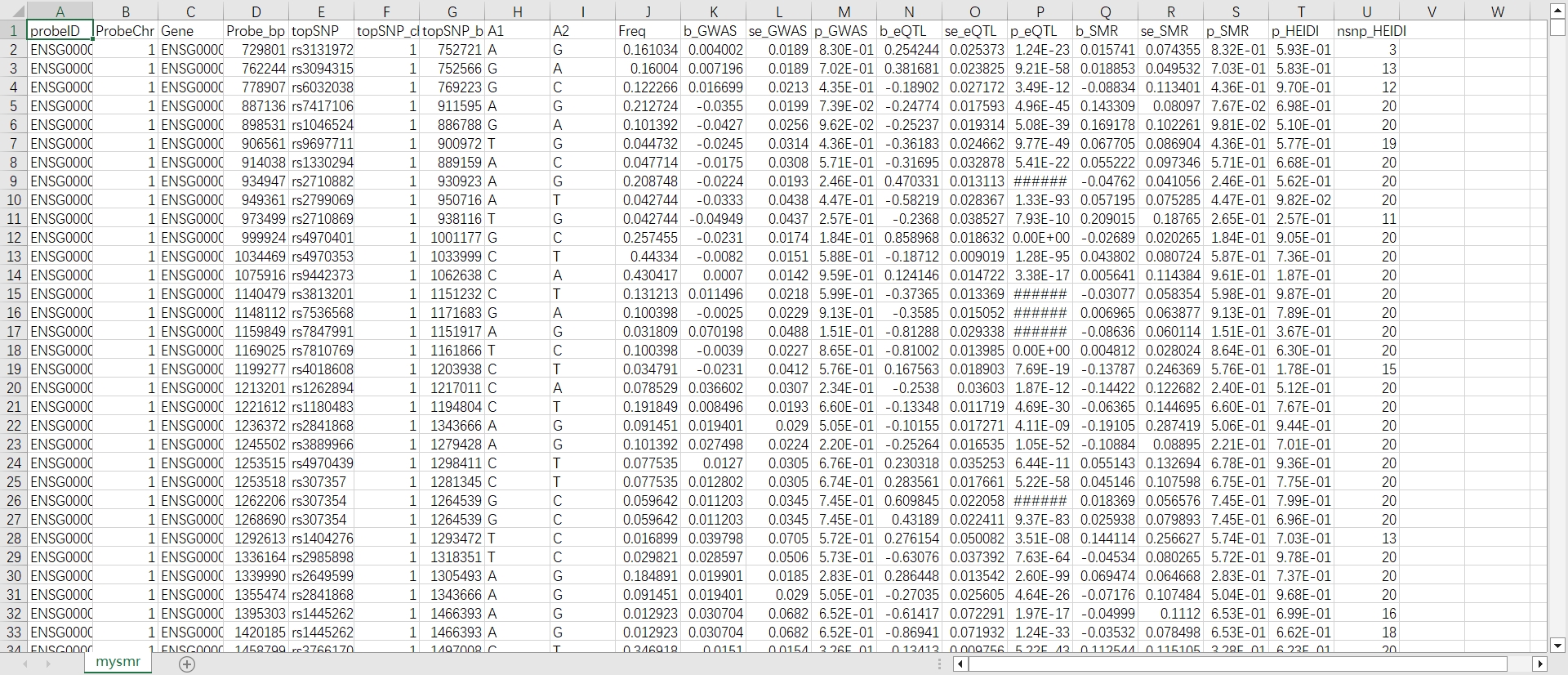
最后我们可以得到一个smr后缀的文件。可以直接使用excel打开,或者复制以后更改成xls格式打开。
具体的结果分析,主要关注SMR的P值,和HEIDI检验的P值。详细分析过程,可以多看看官网提供的论文。
以上就是SMR分析的整个过程,除此之外SMR还有可以进行其他的分析。希望大家积极探索发现。
是杨剑教授不是杨健教授
不好意思?,谢谢提醒!